IX) Some specific examples of oncogenes.
(About which you can learn much more in this department's course
"The Biology of Cancer")
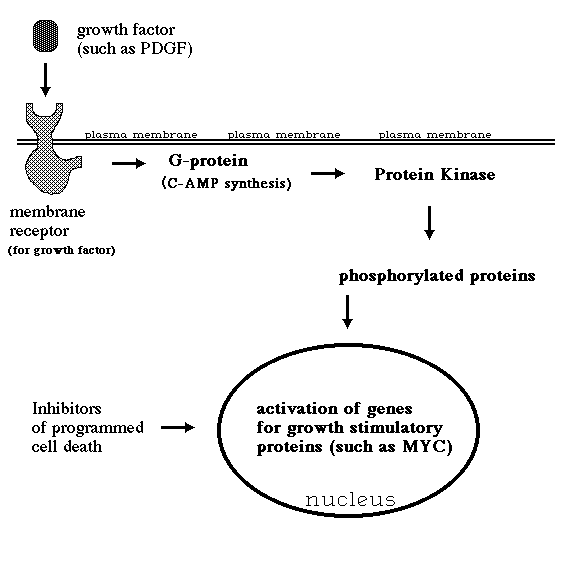
sis: Codes for a form of PDGF (Platelet Derived Growth Factor). PDGF normally serves as a cell-to-cell signal, secreted by platelets and diffusing to other cells such as fibroblasts and smooth muscle cells, which it stimulates to grow and crawl about. If a cell produces its own form of PDGF, it behaves as if it were being constantly exposed to high concentrations of external PDGF; in other words, it stimulates its own growth and locomotion without limit. This is called autocrine stimulation. It is thought that the sis protein binds to the PDGF receptor proteins while these receptors are in the cytoplasm (prior to their insertion into the plasma membrane where their normal interaction with PDGF would have occurred).
erbB: Codes for an abnormal version of the membrane receptor for the extracellular protein called epidermal growth factor. This form of the receptor behaves as if it were constantly bound to molecules of its growth factor, thus constantly sending a false signal stimulating cell growth. A similar oncogene, called erbB-2 seems (based on its base sequence) to code for a receptor for some other (still undiscovered) growth factor. It is found in amplified form in about one fourth of all human breast and ovarian cancers!
ras: The function of its normal equivalent protein is to relay and amplify stimulatory signals, such as those from growth factor receptors. It binds a molecule of GTP whenever it is itself stimulated. It then relays stimulatory signals and continues to do so until its GTP is hydrolysed to GDP. Certain specific amino acid substitutions eliminate this protein's ability to hydrolyze bound GTP, however, so that it remains permanently in its "on" state, constantly "relaying" non-existent signals for the cell to grow and divide. Such mutations of this one oncogene are believed to be responsible for no fewer than one fifth of all human cancers, including up to half of colon carcinomas, and 90% of cancers of the pancreas! (Although note that several different ras genes are known.) Out of 25 average people, ras will kill one of them!
src: Codes for a protein that spontaneously becomes concentrated on the inside surface of the plasma membrane, especially at the sites of cell-substratum adhesion. This protein is an enzyme (a tyrosine kinase) whose effect is to catalyze the covalent bonding of phosphate groups onto the hydroxyl group of tyrosine amino acid residues of proteins .The proteins phosphorylated by the src protein include some which participate in the mechanical linkage between the actin cytoskeleton and materials to which the outside surface of the plasma membrane attaches; these changes may thus be responsible for weakening the cell's adhesiveness. Recent work in Patricia Maness' laboratory in the UNC Biochemistry Department indicates that the normal form of the gene functions in the chemotactic guidance of nerve growth cones. The src protein was the first tyrosine kinase to be discovered. Since all previously discovered kinases (there are many of them in the cell) had catalyzed the bonding of phosphates to the hydroxyl groups of serines and threonines, it was first assumed that phosphorylation of tyrosines was peculiar to cancer cells. But many normal tyrosine kinases have subsequently been found.
myc: Codes for a nuclear protein whose normal function seems to be as some kind of a transcription factor promoting cell growth. For example, when a normal cell is stimulated to grow and divide (for example, by exposure to PDGF), then the c-myc gene product (protein) temporarily increases in concentration; conversely, this gene normally becomes inactive in non-mitotic cell types. Many human cancers have been shown to have undergone amplification of the c-myc gene (often about 10 copies of the gene) this includes many cases of leukemia and about 30% of lung cancers of the highly lethal "small cell" type and breast cancers. The progression of cancerous cells to ever more and more aggressive states is frequently traceable to further amplification of the myc gene.
bcl-2: This name stands for B cell lymphoma and the protein for which this gene codes seems to have the normal function of inhibiting the spontaneous death of B-lymphocytes. In order that the total number of B cells in your body does not continue to increase without limit (because of constant exposure to different antigens) it is essential that the great majority of B-cells self-destruct. This self-destruction phenomenon ("apoptosis") will be discussed more fully below. When too much bcl-2 protein is produced in a given B cell, this blocks the self-destruction. Trans-genic mice with duplications of the bcl-2 gene accumulate abnormal concentrations of lymphocytes, among other abnormalities. Many human lymphomas result from promoter or enhancer sequences of the antibody genes (on the 14th chromosome) accidentally becoming spliced to the site on the 18th chromosome where the bcl-2 gene is located; whenever these B cells "try" to make antibody molecules, what they make instead is lots of the bcl-2 protein. These cells therefore accumulate to form a slow-growing lymphoma (which is always fatal, although not usually until one of the bcl-transformed clones has subsequently also been transformed by an over-activity of the myc oncogene). Prior to this they grow slowly, making it especially paradoxical that these lymphomas can be caused to shrink almost to nothing by the use of growth-poisoning chemotherapeutic drugs.
* Note that most of the oncogenes listed above are part of a "chain of command" by which external signals, especially protein growth factors, normally stimulate cell growth and division. A typical sequence of events would be for a (A) growth factor molecule to diffuse up to a cell's outer surface, then (B) bind to a membrane protein that serves as a specific receptor for that growth factor, with the conformation (C) or other properties of this receptor being changed by the binding, thereby causing either the activation of a cytoplasmic enzyme (that might be a protein kinase or ), or (D) the activation of a g-protein (such as the c-ras protein) that would then (E) activate a protein kinase, which would phosphorylate various cytoplasmic proteins, including some involved in cell adhesion, and would also stimulate increased transcription and translation of (F) genes for certain transcription factors such as c-myc. Paradoxically, myc also tends to stimulate apoptosis (G), unless counteracted by other gene products such as bcl-2.
Cancer cells from actual patients usually contain over-active versions of several different oncogenes, not infrequently 3 or 4 or more. Typically, there is one that acts at the nuclear level (such as myc) and one or more (such as ras or src) that act at the cytoplasmic level. For several years (between about 1982-86) it was confidently believed that cancer wouldn't occur unless there were at least two, and that one of the two had to be cytoplasmic in its action (like sis, erb, ras or src) and that the other had to act at the nuclear level (like myc). But this is no longer believed.
At current rates in the USA, 25 % of you will get cancer.
Four-fifths of this 25% will die of their cancer.
That's 20 people out of every one hundred.
You have to kill just those cells in which... What was it, again?
Until the 1940s, tuberculosis killed as high a percentage of Americans as cancer does now. Then streptomycin was discovered. Guess how it worked.
|